
CLN1 Disease
Are you a genetic carrier for Neuronal Ceroid Lipofuscinosis? Find out with this DNA Test.
- Detects the PPT1 variant linked to CLN1 disease
- Carrier screening test intended for couples who are planning to become pregnant
- Determine if your child is at risk of developing this condition that affects the nervous system
- 100% private and confidential online results
Already have DNA markers? Sign in and upload your data to view results.
Need to take the DNA Test? Order our easy-to-use swab kit.
Detailed Description
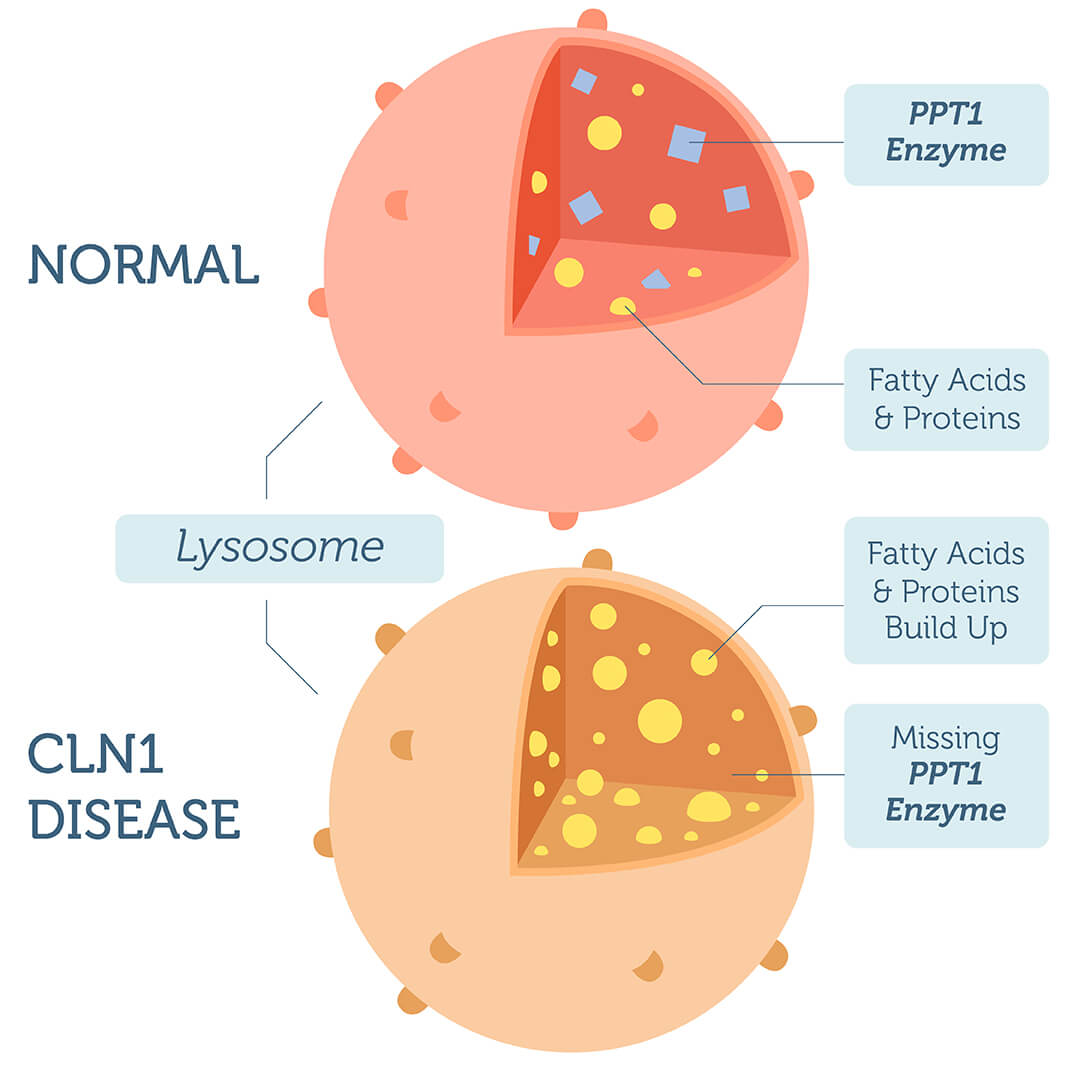
Neuronal ceroid lipofuscinosis (NCL) is a family of inherited genetic diseases that affects the nervous system, caused by the build up of the pigment lipofuscin. Lipofuscin, mainly made up of fats and proteins, is considered to be an “aging” or “wear-and-tear” pigment, as nerve cells, and other organs like the liver, spleen, heart and the kidneys accumulate it over time. However, in affected people there is a severe build up of lipofuscin leading to the symptoms of the disorder.
CLN1 disease is characterized by seizures, vision loss and intellectual disability. It is caused by genetic variation in the PPT1 gene, which gives instructions to make the enzyme palmitoyl-protein thioesterase 1 found in the lysosomes. The Ppt1 enzyme is responsible for removing a specific type of fatty acids from proteins, so they can be broken down. People with PPT1 variants produce a protein with impaired function, which results in the build up of partially broken down fats and proteins in the lysosome.
Take this test to find out whether you are a genetic carrier of PPT1 variants and are at risk of passing it to your children. This is intended to be a carrier screening test for couples who are considering to start a family.
The Genetics
CLN1 disease is caused by genetic variation in the PPT1 gene, which gives instructions to make the enzyme palmitoyl-protein thioesterase 1 involved in the break down of certain proteins. People with CLN1 disease produce an enzyme with impaired function, leading to the build up of partially broken down fats and proteins and cell damage. Those who present with the later-onset cases of CLN1 disease make some functional palmitoyl-protein thioesterase 1 enzyme, allowing the proper break down of some proteins. As the build up of fats and proteins is slower, symptoms appear later in life.
CLN1 disease is inherited in an autosomal recessive pattern, which means two defective copies of the PPT1 gene must be inherited by an affected individual for symptoms to manifest

Variants Tested
This test looks at three variants of PPT1 linked to CLN1 disease observed in 98% of CLN1 cases in people of Finnish descent and 59% of cases in those of Northern and Western European descent.
- rs137852700
- rs137852695
- rs137852696
Understanding your carrier status for these variants will help you understand the risk of passing it to your children.
While it provides a risk analysis for CLN1 disease, it may be necessary to use gene sequencing tests, as additional genetic variants are linked CLN1 disease.
Signs & Symptoms
Affected infants are normal at birth and develop normally until symptoms start to appear around 18 months
- Brain cells die over time resulting in brain atrophy and small head
- Hypotonia (decreased muscle tone)
- Intellectual and motor disability
- Seizures
- Muscle twitches/spasms
- Vision loss (and eventual blindness)
- Feeding difficulties as the symptoms get worse
How It Works
Step 1: Sign up for a free Genebase account.
Step 2: Upload your DNA markers to Genebase.
Step 3: Login to your account to access your results when they are ready.